Projects Modelling Evolutionary Trajectories from Experimental Evolution Studies of Drosophila Species
(with Antti Honkela and Christian Schloetterer) Polymorphism-Aware Phylogenetic Models (with Nicola De Maio) Estimation of Population Genetic Parameters from Pooled Next Generation Sequencing Data
(with Andreas Futschik, Robert Kofler and Christian Schloetterer) Empirical Codon Models
(with Maria Anisimova, Nick Goldman and Ian Holmes) Patterns of Positive Selection on the Mammalian Tree
(with Tomas Vinar, Rute Da Fonseca, Rasmus Nielsen and Adam Siepel)
This project combines NGS technologies with experimental evolution studies. The
substantial decrease in costs has made it feasible to not only
sequence the last generation of a population at the end of a long-term
artificial selection experiment but to
sequence intermediate generations. Using NGS technologies we monitor the allele
frequency changes in populations that undergo a selection experiment
for temperature
adaptation. The resulting data represents evolutionary trajectories, time-series
data, that we analyse using Gaussian Process models. A Gaussian process is a
probability distribution over functions that can represent changes in
concentration.
Analogous to the Gaussian distribution which is fully characterised by
a mean function
and a covariance function. To simplify the inference we will use population
genetic models describing changes in allele frequencies such that they
will allow
us to simplify and reduce the number of parameters of the covariance function. A
further advantage of the Gaussian process approach is that it can
handle replicate
population to identify trends across populations.
Comparative analysis of genomes of related species, and of different
individuals of the same species, can reveal adaptive trends in the
history of the considered taxa, as well as show intensity and genomic
variation of evolutionary patterns of species that undergo speciation.
However, these intra and interspecific data also bring new challenges,
such as the presence of incomplete lineage sorting and ancestral
shared polymorphisms.
We have developed a new method called POlymorphisms-aware phylogenetic
MOdel (PoMo). It is a phylogenetic Markov model with states
representing fixed alleles as well as polymorphisms at different
allele frequencies. A substitution is hereby modeled through a
mutational event followed by a gradual fixation. Polymorphisms can
either be observed in the present (tips of the phylogeny) or be
ancestral (present at inner nodes). With this approach, we naturally
account for incomplete lineage sorting and shared ancestral
polymorphisms. Our method can accurately and time-efficiently estimate
the parameters describing evolutionary patterns for phylogenetic trees
of any shape (species trees, population trees, or any combination of
those). Furthermore, we are able to disentangle the contribution of
mutation rates and fixation biases in shaping substitution patterns.
Pooled sequencing at population level is often cheaper and more
accurate than individual
sequencing. We are developing software which implements the our unbiased
estimates of Tajima’s pi and Watersons theta . We not only account
for the bias derived
by pooled sequencing, but also for the one generated by sequencing
errors, that are
higher in next generation sequencing.
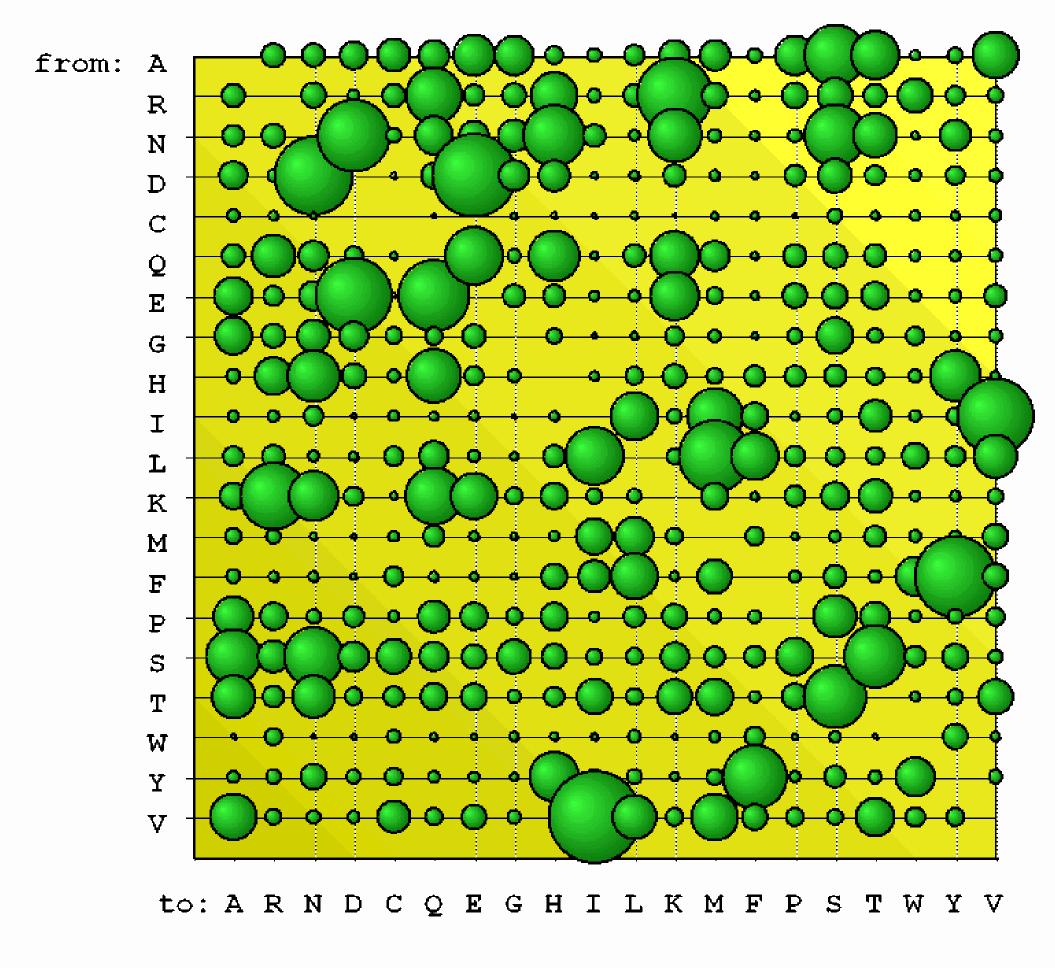
We have estimated a first empirical codon model using maximum likelihood methods. We show that modelling the evolutionary process is improv
ed by allowing for single, double and triple nucleotide changes and that the affiliation between DNA triplets and the amino acid they encode is
a main factor driving evolution. We plan to extend this approach to the estimation of a codon model assuming rate heterogeneity among codon sites caused by the influence of natural selection. Web
page
We have performed the most comprehensive examination of positive selection on the mammalian tree to date, using the six high-coverage genome assemblies now available for eutherian mammals (human, chimpanzee, macaque, mouse, rat, dog). The increased phylogenetic depth of this data set results in substantially improved statistical power, and permits several new lineage- and clade-specific tests to be applied. Webpage